Manufacturing process technology transfer for first-in-human studies is no easy feat. This whitepaper explores how strategic tech transfer planning de-risks the transition from development to GMP manufacturing for first-in-human trials
Introduction
Cell-based therapies hold large potential for curative treatment of many diseases, yet their manufacture is inherently complex. When progressing from early development through to commercial production, one of the most challenging steps is the transfer of the manufacturing process from the development stage to a GMP-manufacturing facility (in many cases, using an external partner) for a first-in-human clinical trial. Especially for small biotech companies, this step is critical as it demands successful cooperation between both companies over several years, with substantial financial and personnel resource commitments. To help early developers navigate these manufacturing transitions, this whitepaper explores the key role of technology transfer activities. Dive into some of the main challenges and considerations, and the steps required to ensure the successful transfer of product and process knowledge between teams and facilities to support a first-in-human study.
What is technology transfer, and how is it involved in the drug product lifecycle?
Technology transfer, often referred to as “tech transfer”, is an essential part of drug development and manufacturing. It involves the transfer of the manufacturing process to make the cell-based product and all necessary analytical methods from the biotech company (Sending Unit, (SU)) to the external partner (Receiving Unit, (RU)), which is typically a contract development and manufacturing organization (CDMO). A successful tech transfer ensures that the manufacturing and analytical processes are faithfully reproduced at the RU, maintaining consistency with the SU’s procedures. It aims to safeguard product quality, ensure alignment with regulatory filings such as clinical trial applications or marketing authorizations, and meet all applicable GMP and other regulatory requirements.
According to the ICH guideline Q10 on pharmaceutical quality system,
"The goal of tech transfer activities is to transfer product and process knowledge between development and manufacturing, and within or between manufacturing sites to achieve product realization. This knowledge forms the basis for the manufacturing process, control strategy, process validation approach and ongoing continual improvement."
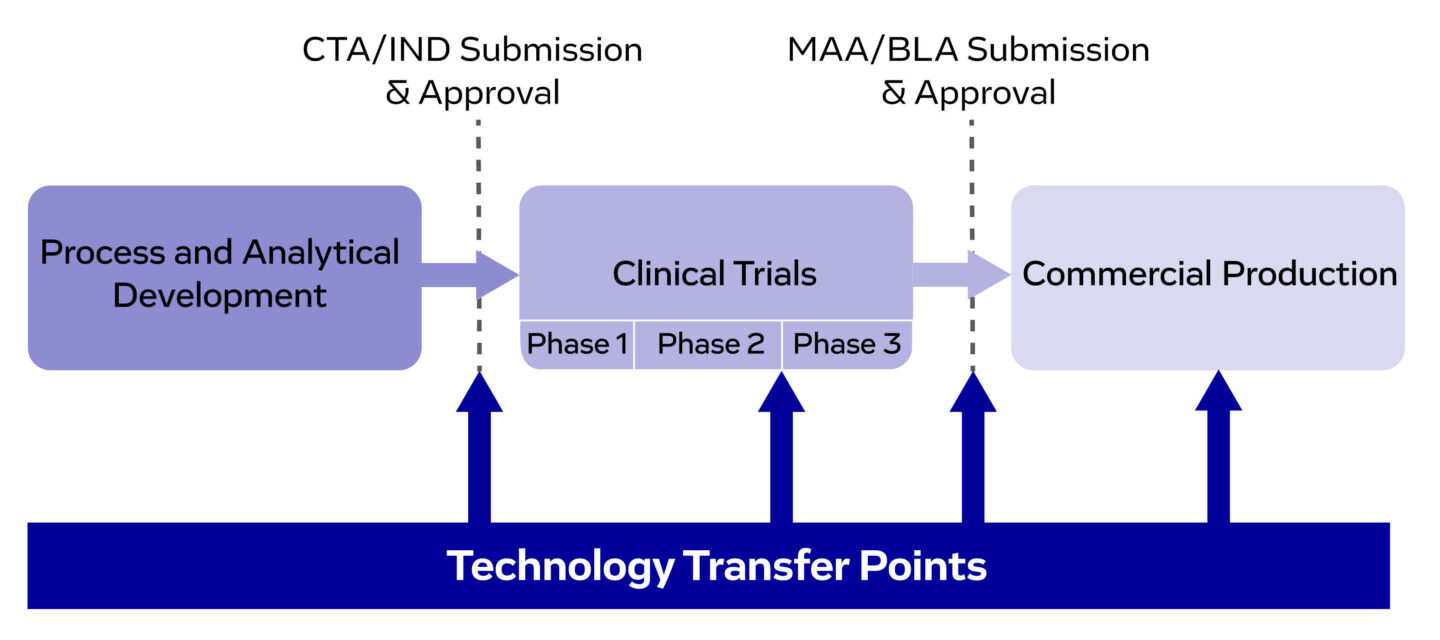
Figure 1. Technology transfer points throughout drug development and manufacturing, ensuring a smooth progression from preclinical development to commercial production.
Tech transfer is critical throughout several stages of the drug product’s lifecycle. It enables the successful transition from development to GMP manufacturing for initial clinical supply. Additional transfers may be required during later development phases and market entry, to scale product supply or to offer the product in different regulatory regions.
What are the specific challenges faced during the tech transfer of a cell therapy product?
Developers often face significant challenges when transferring manufacturing processes for cell-based products between organizations. These include maintaining product quality while adapting processes, and ensuring regulatory compliance where required. A common issue is the use of research-grade raw materials, as higher-quality alternatives remain scarce. This may necessitate material substitutions during tech transfer or the implementation of additional testing to ensure consistent quality.
Standardized platform processes are still limited, leading to the use of diverse equipment across developers. As a result, tech transfer may require switching to a different device at the CDMO or integrating a new one. Both options involve considerable effort. Additionally, cell-based products often rely on unique analytical assays, which can be difficult to implement and validate at the RU.
Furthermore, cell therapy manufacturing often relies on manual processing steps, which may require significantly extended time in a cleanroom environment compared to operations in a development laboratory. This increased processing time may necessitate a reassessment of the in-process stability of cell-based intermediates, to ensure quality and viability are maintained throughout the extended handling period.
Given these complexities, tech transfer in cell therapy demands substantial time, labor, materials, and cost. Ensuring a successful transfer is critical, as poor execution can compromise product quality and patient safety. To support developers in navigating these challenges, we’ve outlined the key steps for a successful tech transfer, from initial planning through to completion.
Key steps to successful cell therapy tech transfer
1. Preparation is the blueprint of success – Tech transfer begins at the sending unit
The sending unit should clearly define the scope of the tech transfer, outlining its regulatory, clinical, and commercial objectives. This includes the consideration of clinical trial timelines, biosafety requirements for handling living cells and tissues, in addition to the geographical regions where the product is intended to be marketed, and the regulatory pathways required.
Also, before initiating any transfer activities, it is essential to have formal agreements in place between the SU and RU. These typically include a Master Service Agreement (MSA), Statement of Work (SOW), and a Quality Assurance Agreement (QAA). The MSA specifies the general terms and conditions (including legal, commercial, confidential, financial, and intellectual property ). The SOW defines the specific scope, tasks, deliverables, timelines, and costs of the transfer, and the QAA outlines the responsibilities and expectations of each party, in terms of GMP and regulatory compliance. Several rounds of review and negotiation are often required for these agreements, which should be considered when planning timelines. It is also advisable to have the agreements reviewed by legal professionals and the relevant SMEs to ensure clarity and flexibility. Also, while reviewing the agreements, the potential for future adaptation and improvement should be a key consideration to help the transfer proceed as smoothly as possible.
2. Initiation - Strategy development and kick-off meetings
Once the scope is established, the transfer process can begin. This starts with initiation to align all stakeholders on the key objectives, timelines, and expectations. During this phase, a robust project management and communication plan should be developed according to general project management standards. A formal, face-to-face kickoff meeting is also highly recommended. Key participants include the project management team, in addition to members of the SU’s process development, analytical development, quality unit divisions, and the RU’s manufacturing, QC, and QA divisions.
3. Document and knowledge transfer
Knowledge transfer is a cornerstone of any successful tech transfer. At the start of the transfer, documentation provided by the SU is crucial to equip the RU with the knowledge required to replicate and maintain the process under GMP conditions. To avoid a last-minute documentation burden, the SU should implement administrative processes that ensure key documents are created and continuously updated throughout development. This proactive approach significantly reduces the workload and risk of gaps when tech transfer activities begin. These documents must clearly outline process workflows and analytical test execution in a format that is both understandable and actionable for the RU. It is also crucial that the SU has identified potential critical quality attributes (pCQA) and process parameters (pCPP) with acceptable ranges during development, as outlined in ICH Q8(R2) guidelines. Further guidance on process and analytical development for cell therapies can be found in our whitepaper. The information obtained from this development phase will help the RU to maintain product quality even if process adaptations need to be implemented, which in many cases is unavoidable when transferring a process to a GMP facility.
All documentation provided by SU should be version-controlled to ensure traceability and transparency. This allows both parties to track the evolution of shared information, minimizing the risk of miscommunication or outdated guidance. Below is an overview of the documentation required:
Development history
- All relevant data from the research and development of the cell-based product to date, including formulation and stability studies, and any preclinical and clinical studies that have been conducted
Process documentation
- A full process description, including all unit operations and critical steps
- Standard Operating Procedures (SOPs) for each process step or the batch record
- Process Flow Diagram (PFD) to visually represent the entire manufacturing process
Analytical documentation
- Analytical SOPs for product characterization, release testing, and stability testing, with a complete list of the materials used
- Reports on method development and pre-qualification to guide method validation
Control strategy documentation
- Including details of the CQAs (e.g. cell identity, viability, purity, potency) and CPPs (e.g. temperature, pH, media composition, incubation time, cell density)
Materials and equipment requirements
- A comprehensive list of all materials used in manufacturing, with specifications and sourcing information (including raw and starting materials, reagents, and consumables)
- Equipment requirements, including the make, model, etc.
- Facility requirements, e.g. required cleanroom classes or design, biosafety level
4. Tech transfer plan, fit assessment, and parallel activities
After document and knowledge transfer, the RU performs a gap analysis and risk assessment based on the information provided by the SU. This will evaluate any risks, and identify any new materials, equipment, processes, methods, and SOPs that will need to be implemented and qualified. Also, the GMP site needs to understand whether their licenses need to be updated.
Following the gap analysis and risk assessment, the RU and SU typically create a joint tech transfer plan. This detailed plan acts as a roadmap for executing and closing the transfer. The plan should clearly articulate what is being transferred (e.g. a clearly delineated manufacturing process, the number and type of analytics, and documentation). It should outline all activities needed to close identified gaps and define the success criteria of each activity (e.g. training for the process and methods, method qualification targets, number of engineering runs and their expected outcomes, etc.). As many of the activities need to be conducted in parallel, it is important to generate a detailed list of tasks, responsibilities, and milestones.
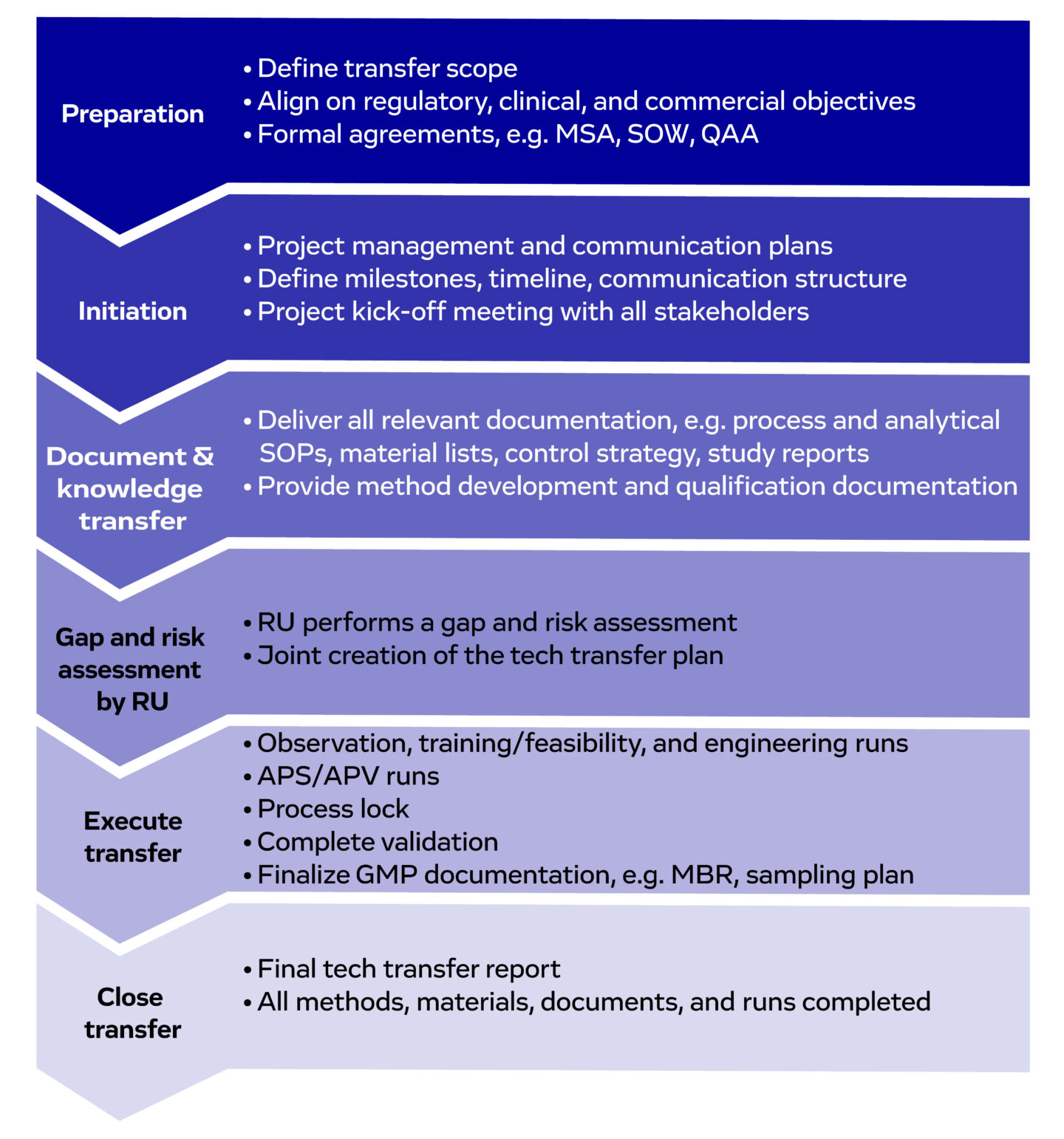
Figure 2. An overview of the key stages of cell therapy tech transfer, and the activities involved. Abbreviations: MSA (Master Service Agreement), SOW (Statement of Work), QAA (Quality Assurance Agreement), SOP (Standard Operating Procedure), RU (Receiving Unit). APS/APV (Aseptic Process Validation/Simulation), GMP (Good Manufacturing Practices), MBR (Master Batch Record).
5. Executing the transfer – From training runs through to GMP manufacturing runs
The execution starts with the observation runs at the SU. These give representatives from the RU the opportunity to observe operations in real time, helping them gain a deeper understanding of the manufacturing process, including release testing and in-process controls, sampling strategies, material handling, equipment operation, and facility layout. Following observation runs, training runs / feasibility runs at the RU are carried out, guided by SMEs from the SU. These are practice runs of the full manufacturing process, to help personnel familiarize themselves with critical process steps, such as cell isolation and processing, and final formulation/fill-finish, using equipment and materials specific to their facility. These practice runs should also be used to train the analytical staff in sampling and execution of all required methods. In addition to staff training, these runs build confidence in the execution of the transfer, reducing the risk of deviations, and supporting the readiness of the facility.
After the training runs have been completed, engineering runs are conducted at the RU. These are executed under near-final conditions, generating material for stability studies, GLP toxicology studies, and analytical method validation, all of which are essential for IND-enabling activities. Engineering runs are especially important for confirming that the product can be made according to specifications.
Following the engineering runs, three aseptic process validation/simulation (APV/APS) runs are executed. These runs are required to show that aseptic conditions can be maintained throughout the manufacturing process. Based on observations during the APV/APS runs, final adaptations of the Master Batch Record (MBR) may be implemented, and the process is locked subsequently in a GMP-compliant format. Additionally, a detailed sampling plan is developed, covering the required samples for in-process, release, and stability testing. These activities are critical to ensuring that the RU is fully prepared to manufacture clinical- or commercial-grade material under regulatory compliance, with all quality systems, documentation, and personnel training in place.
6. Closing the transfer
Cell therapy tech transfer can be closed when the RU is ready to manufacture the GMP-compliant cell-based product. This includes the completion of all activities outlined in the tech transfer plan and the resolution of any gaps identified during the gap analysis. Engineering runs should be finalized, and all relevant analytical methods must be appropriately qualified or validated. Additionally, qualification of all raw materials and devices must be concluded, and all GMP-relevant documentation must be completed, reviewed, and approved. Deviations must be closed. Following this, the transfer is formally closed with the completion of a comprehensive Tech Transfer Report, which documents the entire process, acceptance criteria, outcomes, and readiness for clinical/commercial manufacturing, explaining and justifying any deviations.
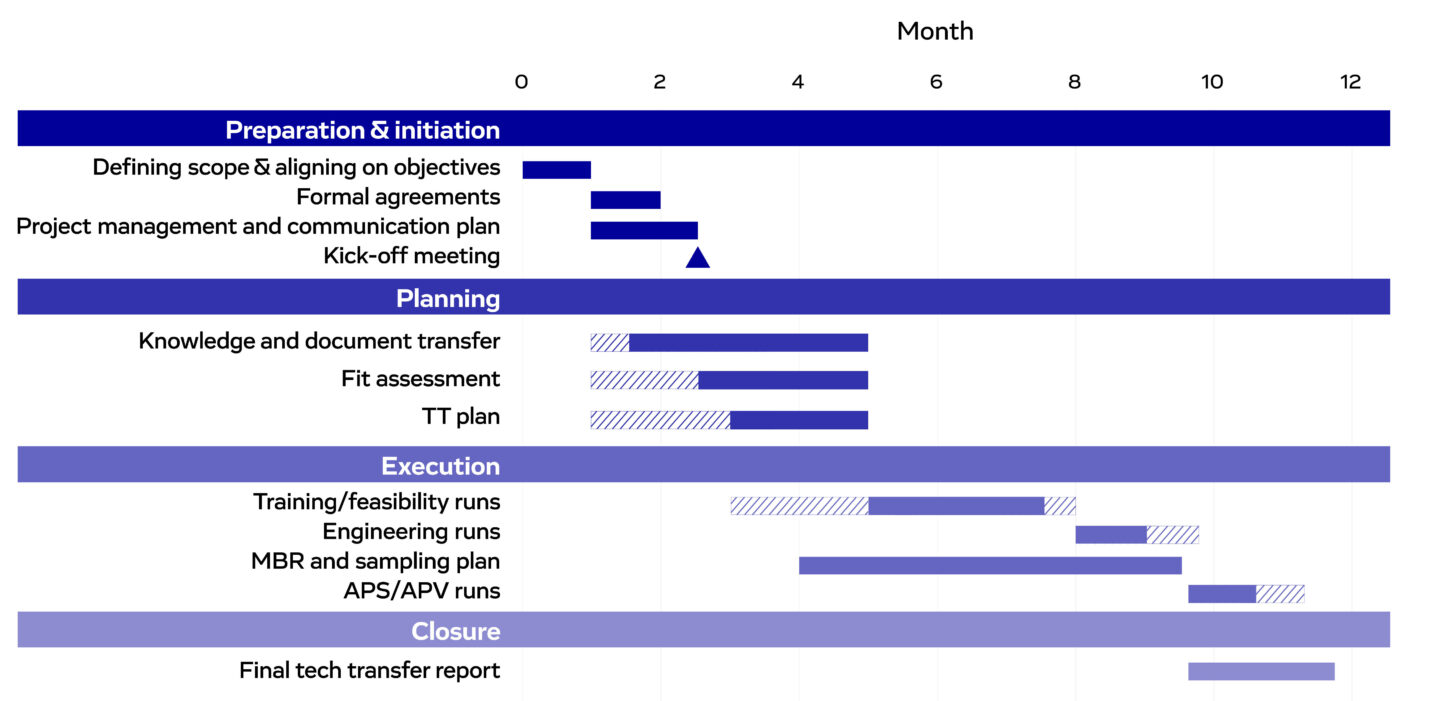
Figure 3. An example Gantt chart for cell therapy manufacturing tech transfer. Hatched bars represent stages that may commence earlier or require extended durations, depending on project needs.
Given the complexity of cell therapy tech transfer, how can you ensure success?
As described in this guide, tech transfer is a vital yet complex part of the cell therapy product lifecycle. Many steps are needed to progress from initial scoping through to completion, each requiring meticulous planning and documentation to consistently replicate the manufacturing process. To ensure success, the seamless alignment of functional teams is required, including manufacturing, QC, QA, regulatory affairs, and project management. Each team plays a fundamental role in the transfer, with misalignments potentially causing unpredicted changes to the manufacturing process, risking the quality and safety of cell-based products.
Given the risks and complexity, having the right expertise, experience, and capabilities is critical for success. This makes choosing a Contract, Development, and Manufacturing Organization (CDMO) a key decision that could shape the outcomes of your tech transfer. Several considerations should guide CDMO selection, including compatibility with your regulatory, development, and commercial goals (e.g. requirements for regulatory compliance, operational logistics, biosafety, and market-entry). Geographical location is also an important consideration, as selecting a CDMO in the same regulatory region can simplify compliance. Additionally, geographical proximity to the anticipated clinical trial site can reduce the risks associated with the shipping and handling of the sensitive cell-based products.
A structured selection process is required to ensure you have selected the right CDMO for your tech transfer. This typically begins with gathering basic information through a Request for Information (RFI) and initial discussions to assess general suitability. This is followed by the development of a robust Request for Proposal (RFP), outlining the high-level scope, expectations, timelines, and regulatory requirements. Alongside RFP evaluation, considering the CDMO’s track record and regulatory readiness is crucial. Once a shortlist is created, site visits can help verify capabilities and ensure alignment on expectations.
When transferring the manufacturing of cell-based products, sponsors need a trusted partner. Evotec is a proven leader, with specialized capabilities in iPSC-based, autologous, and allogeneic therapies, and deep expertise across a range of disease areas and therapeutic modalities. We offer a fully integrated platform that brings all cell therapy development and manufacturing capabilities under one roof – from project inception through to early and late-stage clinical development. This includes in-house clinical operations, advanced GMP manufacturing, rigorous quality control systems, and comprehensive regulatory know-how. Our integrated approach ultimately ensures greater cross-functional alignment throughout the full transfer process, minimizing the risks of deviations and facilitating smoother transitions to GMP manufacturing.

Figure 4. An overview of the steps required to select the most appropriate CDMO for cell therapy tech transfer.